Gel electrophoresis is a procedure used to separate biological molecules by size. The separation of these molecules is achieved by placing them in a gel with small pores and creating an electric field across the gel. The molecules will move faster or slower based on their size and electric charge.
The process of gel electrophoresis works because negatively charged molecules move away from the negative pole of the electric current and smaller molecules will move faster than larger molecules. Thus, a size separation is achieved within the pool of molecules running through the gel. The gel works in a similar manner to a sieve separating particles by size. The electrophoresis works to move the particles, using their inherent electric charge, through the sieve.
When researchers are trying to distinguish between different segments of DNA, for example, the process is simple. The samples are loaded into channels at the start of the gel. Each DNA molecule has the same charge (-1), because DNA is formed by the same 4 nucleotides and always carries a slightly negative charge regardless of its size. Therefore, each DNA molecule will have the same force pulling it through the gel.
However, the size of each molecule hinders its progress through the gel. Large molecules hit parts of the gel matrix, and are slowed down. Small DNA molecules can slip between the various components of the gel matrix, and quickly make their way to the other side of the gel. After a certain amount of time, the dyed DNA molecules can be seen aggregating in different areas of the gel, based on how far they moved during gel electrophoresis. This allows researchers to identify the segments, and compare the DNA of different organisms.
The purpose of gel electrophoresis is to visualize, identify and distinguish molecules that have been processed by a previous method such as PCR, enzymatic digestion or an experimental condition. Often, mixtures of nucleic acids or proteins that are collected from a previous experiment/method are run through gel electrophoresis to determine the identity or differentiate between molecules.
The broad steps involved in a common DNA gel electrophoresis protocol:
The DNA is isolated and preprocessed (e.g. PCR, enzymatic digestion) and made up in solution with some basic blue dye to help visualize the movement of the sample through the gel.
TAE buffer provides a source of ions for setting up the electric field during electrophoresis. The weight-to-volume concentration of agarose in TAE buffer is used to prepare the solution. For example, if a 1% agarose gel is required, 1g of agarose is added to 100mL of TAE. The agarose percentage used is determined by how big or small the DNA is expected to be. If one is looking at separating a pool of smaller size DNA bands (<500bp), a higher percentage agarose gel (>1%) is prepared. The higher percentage of agarose creates a denser sieve to increase the separation of small DNA length differences. The agarose-TAE solution is heated to dissolve the agarose.
The agarose TAE solution is poured into a casting tray that, once the gel solution has cooled down and solidified, creates a gel slab with a row of wells at the top.
The solid gel is placed into a chamber filled with TAE buffer. The gel is positioned so that the chamber wells are closest to the negative electrode of the chamber.
The gel chamber wells are loaded with the DNA samples and usually, a DNA ladder is also loaded as reference for sizes.
The negative and positive leads are connected to the chamber and to a power supply where the voltage is set. Turning on the power supply sets up the electric field and the negatively charged DNA samples will start to migrate through the gel and away from the negative electrode towards the positive.
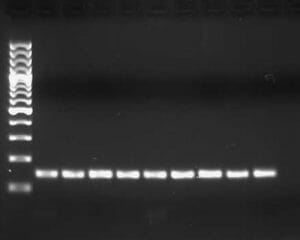
Once the blue dye in the DNA samples has migrated through the gel far enough, the power supply is turned off and the gel is removed and placed into an ethidium bromide solution. Ethidium bromide intercalates between DNA and is visible in UV light. Sometimes ethidium bromide is added directly to the agarose gel solution in step 2. The ethidium bromide stained gel is then exposed to UV light and a picture is taken. DNA bands are visualized in from each lane corresponding to a chamber well. The DNA ladder that was loaded is also visualized and the length of the DNA bands can be estimated. An example is given in the figure below.
There are two types of gel electrophoresis: native and denaturing. Native gel electrophoresis usually attempts to keep RNA or protein in its native structure while running it through the gel. Denaturing gel electrophoresis attempts to reduce the RNA or protein into its most linear structure before or during gel electrophoresis.
The denaturation of the RNA or protein is accomplished by adding a reducing agent to the sample, gel and/or buffer. The reducing agent separates bonds within the RNA or protein molecule and thereby reduces its secondary structure. The secondary structure of a protein or RNA will influence, in a non-linear manner, how fast it migrates through a gel. A denatured, linear form of RNA or protein, however, will migrate proportionally to its linear size (base pairs or kilo Daltons). Denaturing gel electrophoresis is often more accurate for size identification, whereas native gel electrophoresis is usually used to identify larger protein complexes.